Рассказываем о том, как в лаборатории разрабатываются методики анализа примесей
Содержание
- Идеология
- Выбор стратегии
- Разработка условий разделения и идентификация примесей
- Предвалидация
- Установление критериев пригодности хроматографической системы
- Оформление отчета о разработке
- Воспроизведение методики на промышленной площадке
- Валидация методики
- Внедрение методики на промышленной площадке
Идеология
Содержание примесей в фармацевтических субстанциях и лекарственных препаратах как правило определяют методом жидкостной хроматографии (ВЭЖХ)
Подходы к нормированию посторонних примесей изложены в руководствах ICH Q3 (в русском переводе). В книге Валидация методик в фармацевтическом анализе (Й. Эрмер, изд. группа компаний ВИАЛЕК , 2013) помимо подходов описаны еще и примеры из реальной практики
В качестве примесей могут выступать исходные компоненты синтеза, полупродукты, продукты разложения (деструкции).
Когда ведется разработка дженерика, потенциальные примеси обычно описаны, и во многих случаях их можно приобрести - это существенно облегчает задачу.
Процесс разработки можно изобразить в виде схемы
 открыть в полном размере
открыть в полном размере
Выбор стратегии
Разработка методики начинается с изучения англоязычной научной литературы - какие примеси для данной субстанции описаны, какими методами они определялись. Это нужно чтобы не пропустить какую-то потенциально важную примесь. Каждый год проводится множество исследований, публикуются новые данные по казалось бы давно известным препаратам.
В нашем университете есть доступ к международным базам научных публикаций (ScienceDirect, базы ACS, SpringerLink и др.), поэтому такой поиск обычно выполняется оперативно. Так же изучаем, какие примеси нормирует производитель в НД на субстанцию, что говорят по этому поводу Американская и Европейская фармакопеи. Если работаем с субстанцией, смотрим технологию - примесями могут оказаться компоненты с предшествующих стадий синтеза.
Чтобы приступить к разработке методики - нужно иметь образцы потенциальных примесей. Поэтому следующим шагом ищем поставщиков: если существуют стандартные образцы примесей USP и EP, выбираем их, если нет - смотрим возможность поставки у компаний, специализирующихся на заказном тонком орг. синтезе (Santa Cruz Biotechnology, Pharmaffiliates и др.)
 Стандарты примесей и основного вещества для разработки одной из методик
Стандарты примесей и основного вещества для разработки одной из методик
Если какие-то примеси недоступны, или речь идет об новых молекулах, то совместно с заказчиком принимаем решение об аттестации стандартного образца предприятия. Заказчик выделяет или нарабатывает образец примеси, а мы подтверждаем его структуру первичными методами: спектроскопией ЯМР, газовой и жидкостной хромато-масс-спектрометрией
Разработка условий разделения и идентификация примесей
Исходя из оптических свойств примесей и основного вещества выбираем длинну волны детектирования (или принимаем решение использовать рефрактометрический детектор).
Далее решается ключевая и самая сложная задача - разработка условий хроматографического разделения. Исходя из свойств разделяемых соединений выбираем хроматографический режим (ОФ, НФ, HILIC, ионный, смешанный) и подбираем хроматографическую систему (конкретную неподвижную фазу, размер зерна сорбента и длину колонки, состав подвижной фазы и режим элюирования), которые обеспечивают полное разделение действующего вещества и примесей. Управля селективностью и удерживанием, стараемся добиться баланса между разрешением и временем анализа. Стараемся добиваться не минимального разрешения, требуемого фармакопеей (Rs=2), а делать «запас», на случай старения колонки при проведении потокового контроля на заводе. Проверяем, не мешают ли определению вспомогательные компоненты препарата (т.н. «плацебо») и системные пики. Естественно, разрабатываемое разделение должно быть устойчивым (робастным).
 Константин Сычев на школе по ВЭЖХ 2017 рассказывает, как разрешение связано с селективностью, удерживанием и эффективностью
Константин Сычев на школе по ВЭЖХ 2017 рассказывает, как разрешение связано с селективностью, удерживанием и эффективностью
Обязательно проводяться стресс-тесты: термическое воздействие, кислотный и щелочной гидролиз, фото-деградация. Смотрим, позволяет ли наша хроматографическая система определить продукты распада (даже если их в "свежем" препарате не наблюдается). При необходимости дорабатываем хроматографическое разделение.
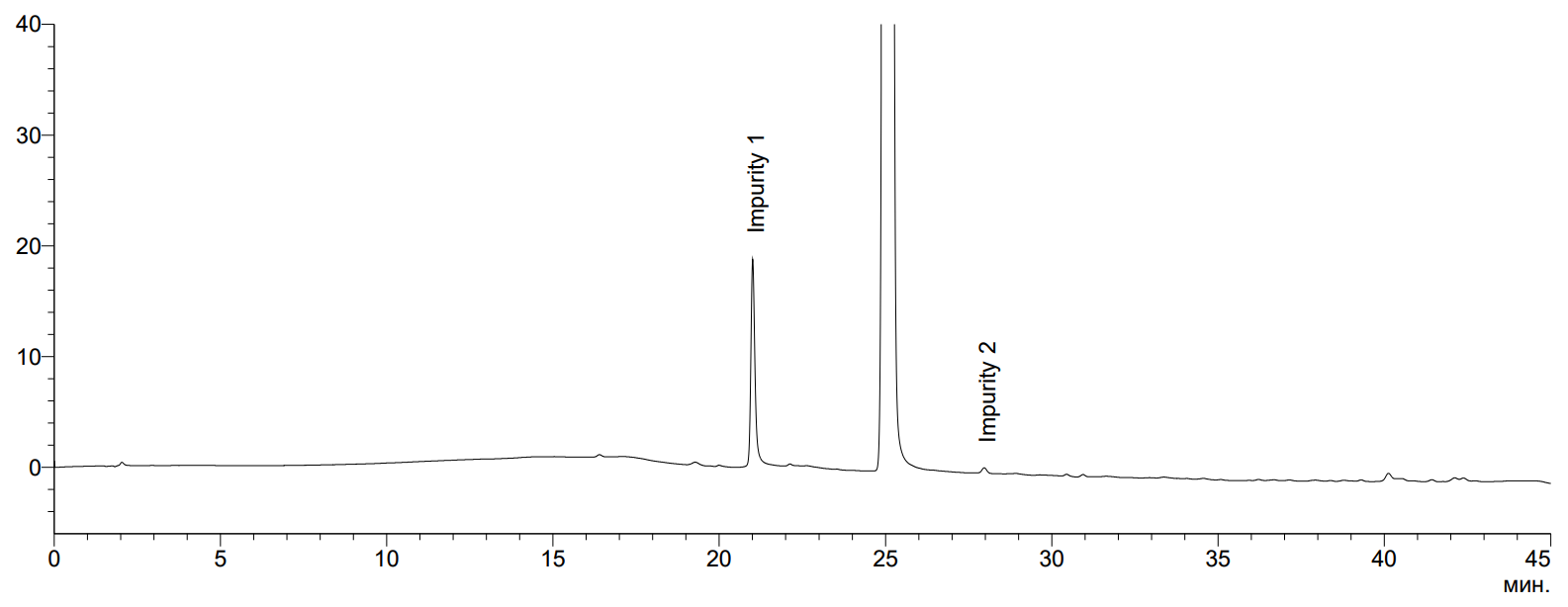
Дальше проводится анализ лабораторных и экспериментальных серий перпарата. Некоторые примеси-деграданты проявляются в ходе "старения" препарата, при этом в субстанции и "свежем" препарате они могут не обнаруживаться. Поэтому при идентификации примесей анализируются образцы "состаренного" перпарата (как правило в условиях ускорренного старения). Проводим идентификацию примесей, имея на руках образцы потенциальных примесей и методом жидкостной хромато-масс-спектрометриии высокого разрешения.
Предвалидация
Следующим шагом идет т.н. «предвалидация», в ходе которой оцениваем метрологические характеристики методики:
- предел количественного определения
- линейность
- прецизионность (повторяемость и промежуточная прецизионность)
- правильность
Это обязательная и очень важная часть разработки. Только проверив метрологические характеристики (пусть и в сокращенном объеме) можно быть уверенными в работоспособности методики. На этом этапе анализ проводится независимо двумя специалистами. Полученные данные позволяют так же обосновать валидационные критерии и критерии пригодности
Критерии пригодности хроматографической системы
После разработки условий разделения и предвалиации, определяемся с критериями пригодности хроматографической системы. В критерии включаем требования к:
- разрешению (с обоснованием выбора критической пары)
- чувствительности (анализ раствора с концентрацией на уровне предела количественного определения)
- xроматографической эффективности
- повторяемости
Отчет о разработке
По результатам разработки оформляется научно-технический отчет, и сама методика
 Отчет о разработке методики анализа
Отчет о разработке методики анализа
Воспроизведение методики на промышленной площадке
Выезжаем на площадку, где совместно со специалистами завода воспроизводим методику, отвечаем на вопросы
Валидация методики
Валидация - экспериментальное доказательство того, что методика позволяет достоверно решать поставленную аналитическую задачу (контролировать качество конкретного препарата или субстанции)
Мы рекомендуем проводить валидацию в заводской лаборатории, которая будет дальше по этой методике вести контроль качества. В этом случае не придется делать верификацию, и специалисты смогут "набить руку" к началу выпуска препарата. Экспериментально обоснованные значения "валидационных критериев" мы приводим в отчете о разработке методики.
Для ряда проектов проводим валидацию на базе нашей лаборатории. План валидации обязательно согласовываем с заказчиком
Внедрение методики в заводской лаборатории
Прежде чем заводская лаборатория начнет проводить анализ по новой методике, ее надо "внедрить"
Внедрение может проходить двумя путями: в форме верификации или трансфера
Вертификация
Верификация – подтверждение лабораторией способности получать достоверные результаты (пригодные для решения конкретной задачи) по готовой валидированной методике
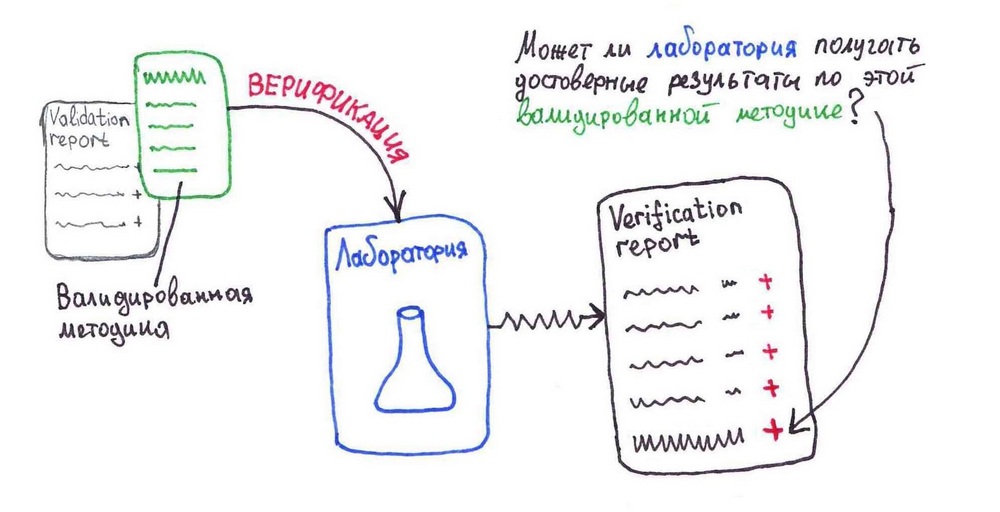
Т.е. верификация проводится заводской лабораторией самостоятельно
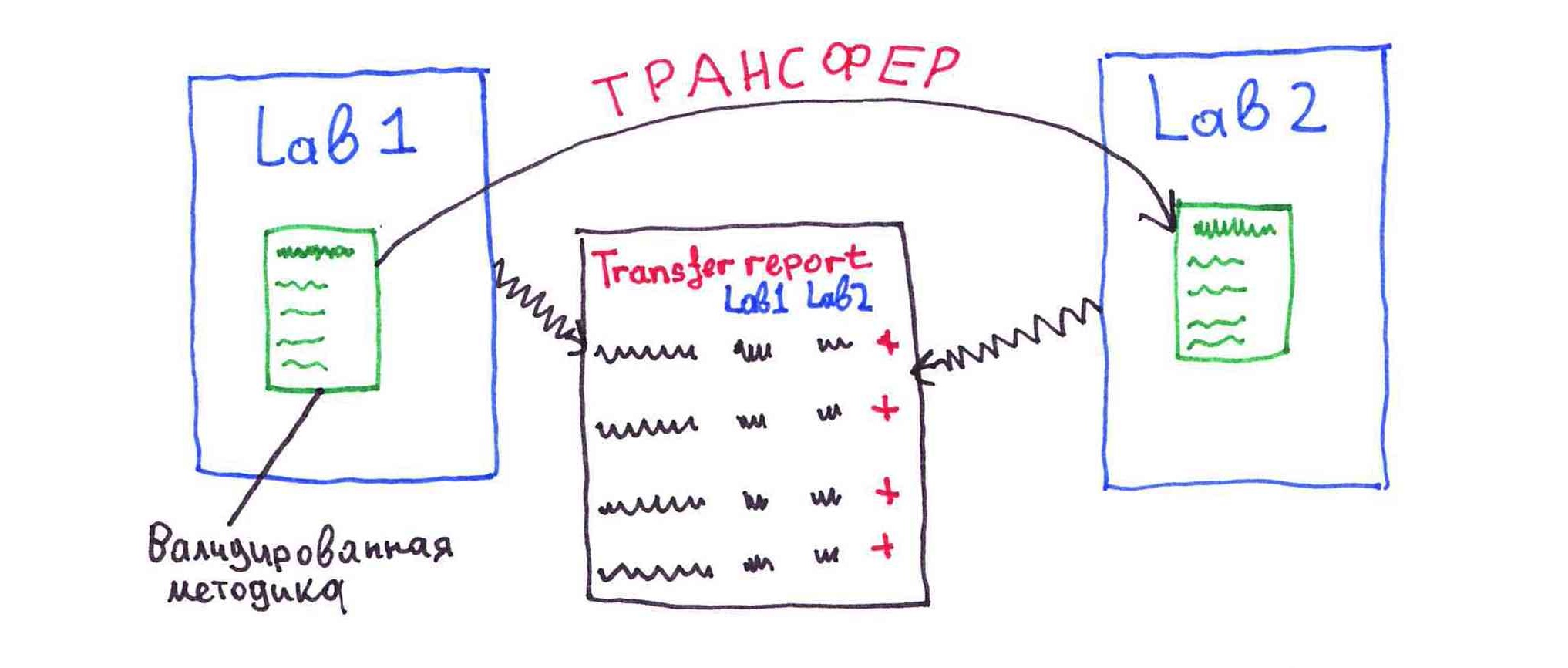
Трансфер
Второй вариант внедрения - в форме трансфера
Трансфер проводится путем проведения сравнительного испытания образца препарата в передающей и принимающей лабораториях. Как и при воспроизведении методики, наши специалисты выезжают на производственную площадку